序章:顯微鏡下的困惑
1970年代初期,在世界各地的腎臟科診室裡,醫師們面對著一個令人困惑的難題。
一名十歲的男孩被帶進診間,他的雙眼腫脹如核桃,腹部膨脹得像懷孕的婦女。驗尿報告顯示他每天流失的蛋白質足以填滿一個茶杯。這是腎病症候群——腎臟過濾屏障崩潰的徵兆。
病理科醫師將腎臟活檢組織置於顯微鏡下,期待看到某種「敵人」的蹤跡——也許是免疫複合物的沉積,也許是感染的痕跡。然而,在螢光顯微鏡下,腎小球閃閃發亮,一切看起來幾乎正常。沒有抗體,沒有補體,沒有明顯的入侵者。
「類脂質腎病」——這是當時的診斷名稱,因為這些孩子的血液中充滿了脂質,而腎臟卻找不到明確的病因。
更奇怪的是,這些患者對類固醇的反應天差地別。有些孩子服用prednisone後,蛋白尿奇蹟般地消失;而另一些孩子即使吞下足以毀掉骨骼的激素劑量,尿液中的蛋白質依然如決堤之水。
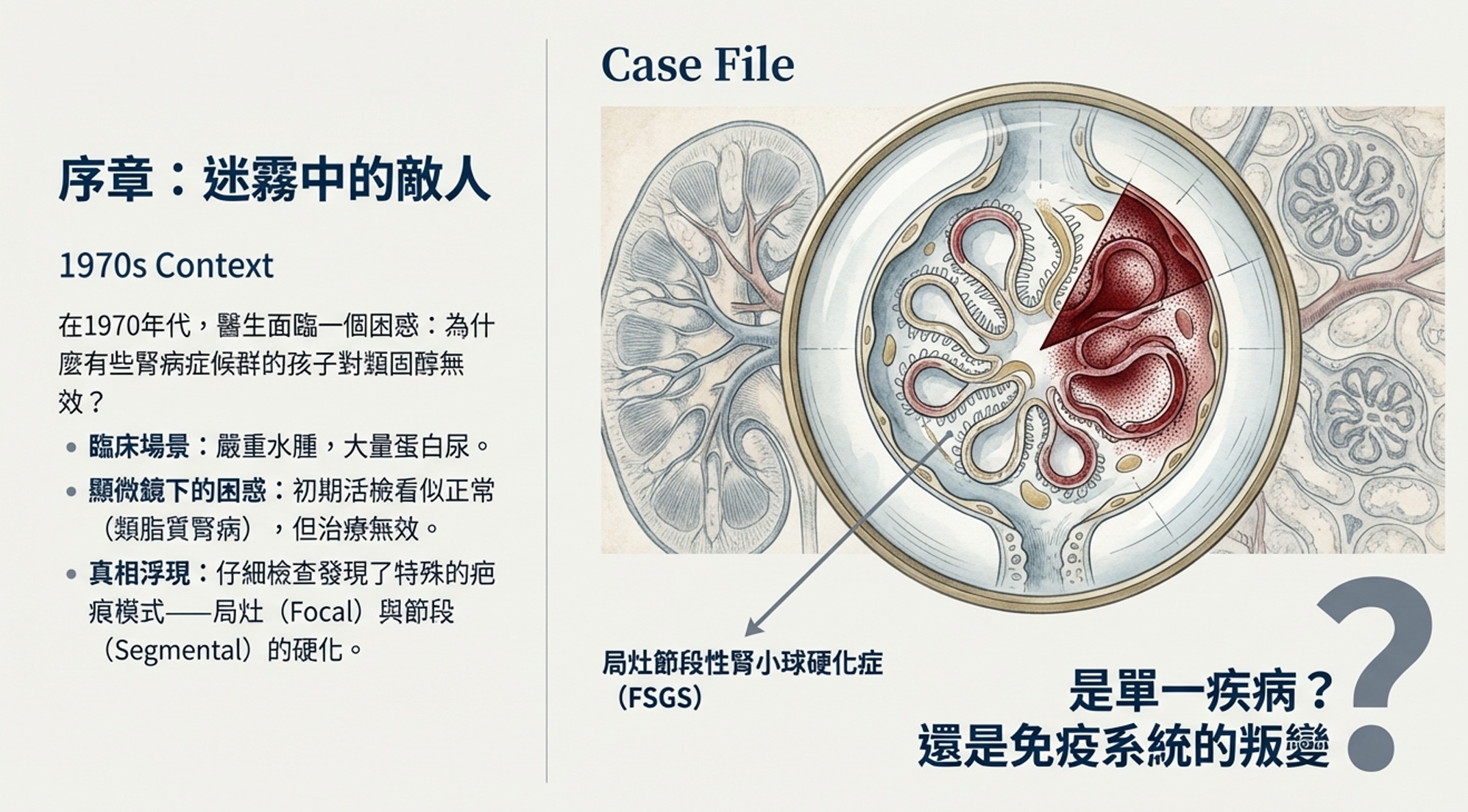
在這些類固醇無效的患者中,病理學家開始注意到一種特殊的病變模式:局灶——只有部分腎小球受累;節段——每個受累腎小球也只有部分區域出現問題。這些區域呈現出一種疤痕化的外觀,毛細血管腔被細胞外基質填塞,有時腎小球絲球會黏附在鮑氏囊壁上。
局灶節段性腎小球硬化症(Focal Segmental Glomerulosclerosis, FSGS)——這個名字就此誕生。但它究竟是什麼?是一種疾病,還是多種疾病的最終共同表現?是免疫系統的叛變,還是腎臟建築的先天缺陷?
這些問題將困擾腎臟病學家半個世紀。
第一部:幽靈因子的假說
第一章:麻疹的啟示(1974)
1974年,美國紐約州立大學下州醫學中心的Robert J. Shalhoub坐在辦公室裡,桌上散落著數十份病歷。作為一名腎臟科醫師,他已經見過太多這樣的案例:孩子們全身水腫,尿液中蛋白質傾瀉而出,卻在腎臟活檢中找不到任何免疫沉積物。
傳統的免疫學思維認為,如果是免疫疾病,就應該看到抗體或免疫複合物的蹤跡。然而這些病人的腎小球在螢光顯微鏡下乾淨得像新生嬰兒的皮膚。
但Shalhoub注意到一個奇特的現象。
「你記得三號床的小病人嗎?」他問他的研究助理。「兩週前還是腎病症候群,現在蛋白尿完全消失了。」
「是因為我們加大了類固醇劑量嗎?」
「不,」Shalhoub搖搖頭,「他得了麻疹。」
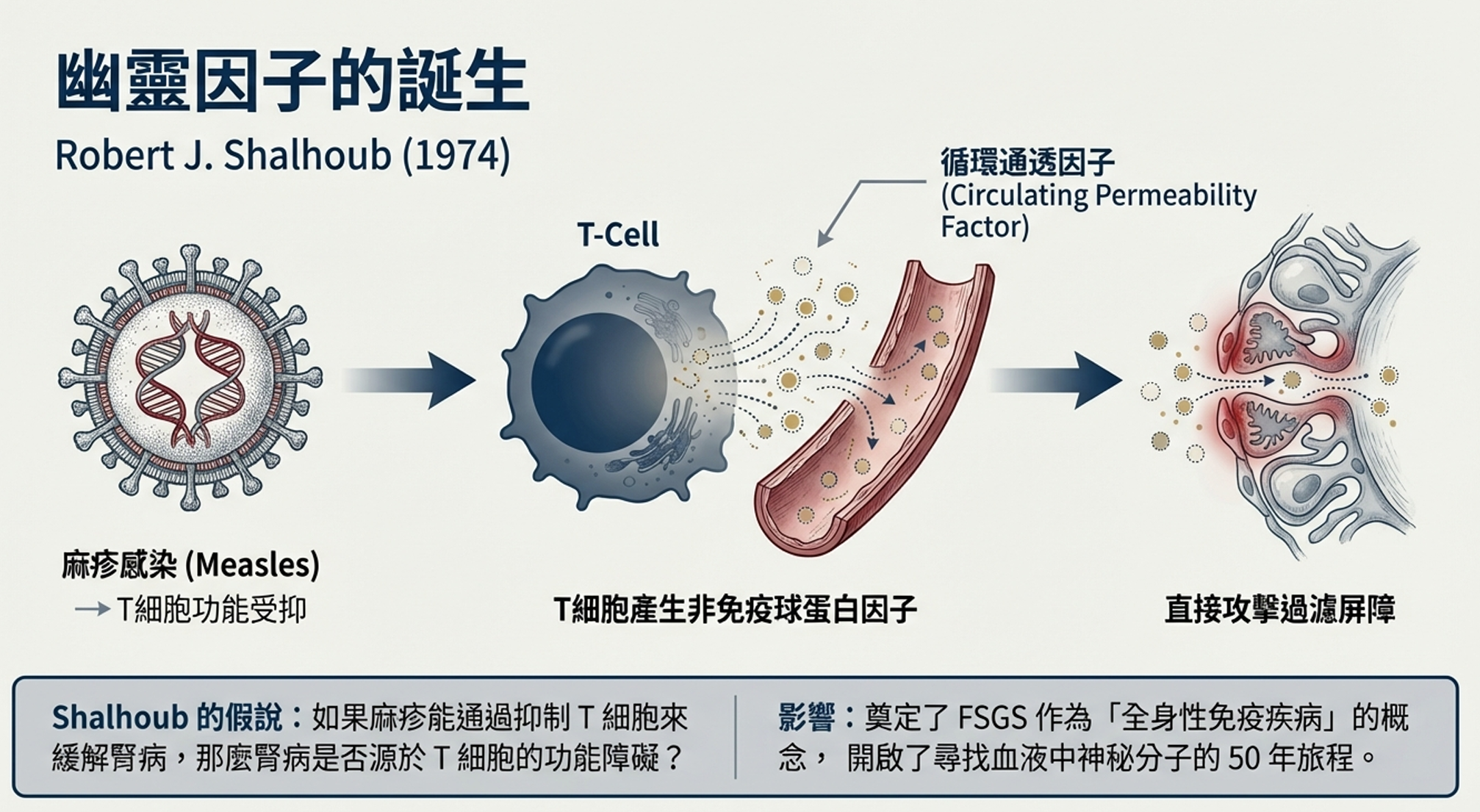
這不是孤例。翻閱文獻,Shalhoub發現多個類似的報告:腎病症候群患者在感染麻疹後自發緩解。麻疹病毒是已知的細胞免疫抑制劑——它會暫時癱瘓T細胞的功能。
一個大膽的假說在Shalhoub腦中成形。
如果問題不是B細胞和抗體呢?如果真正的罪魁禍首是T細胞呢?
他開始構建理論:某個失調的T細胞克隆可能產生一種循環因子——一種非免疫球蛋白性質的淋巴因子——這種因子不會沉積在腎小球上,因此螢光顯微鏡看不到任何東西,但它會直接攻擊腎小球的過濾屏障,使其變得「漏」。
這就解釋了為什麼環磷醯胺(一種強效淋巴細胞抑制劑)對這些患者有效——它殺死了產生毒性因子的T細胞。這也解釋了麻疹的神奇療效——病毒暫時抑制了細胞免疫。
1974年9月,Shalhoub在《刺胳針》(The Lancet)發表了他的假說。論文標題平淡無奇:「類脂質腎病的發病機制:一種T細胞功能障礙」。但這篇僅四頁的論文將成為FSGS研究的「創世紀」。
他提出的「循環通透因子」概念——一種能增加腎小球通透性的神秘血漿分子——將主導此後五十年的研究。科學家們將在血液中搜尋這個幽靈般的因子,提出一個又一個候選分子,然後一次又一次地失望。
但Shalhoub種下的種子已經發芽。腎臟病學家們開始以全新的眼光審視這些患者:也許他們面對的不是腎臟本身的疾病,而是某種系統性免疫失調的腎臟表現。
第二章:瘟疫帶來的新疾病(1984)
十年後,1980年代初,一場新的瘟疫正在紐約肆虐。
人們稱它為AIDS——後天免疫缺乏症候群。這種神秘的疾病摧毀患者的免疫系統,使他們死於各種機會性感染。它主要影響男同性戀者和靜脈吸毒者,在當時的社會引發恐慌和歧視。
在布魯克林的Downstate醫學中心,腎臟科醫師T.K.S. Rao開始注意到一種令人不安的模式。越來越多的AIDS患者被轉介到他的門診,不是因為感染,而是因為他們的腎臟正在快速衰竭。
「這跟我們以前見過的任何東西都不一樣,」Rao告訴他的同事。「他們的蛋白尿非常嚴重——有些人每天流失超過10克蛋白質。而且進展太快了。一個病人從正常腎功能到需要透析,只用了六週。」
Rao和他的團隊開始系統性地研究這個現象。他們檢視了92名AIDS患者的腎臟病變,發現其中11人呈現一種特殊的腎病模式。
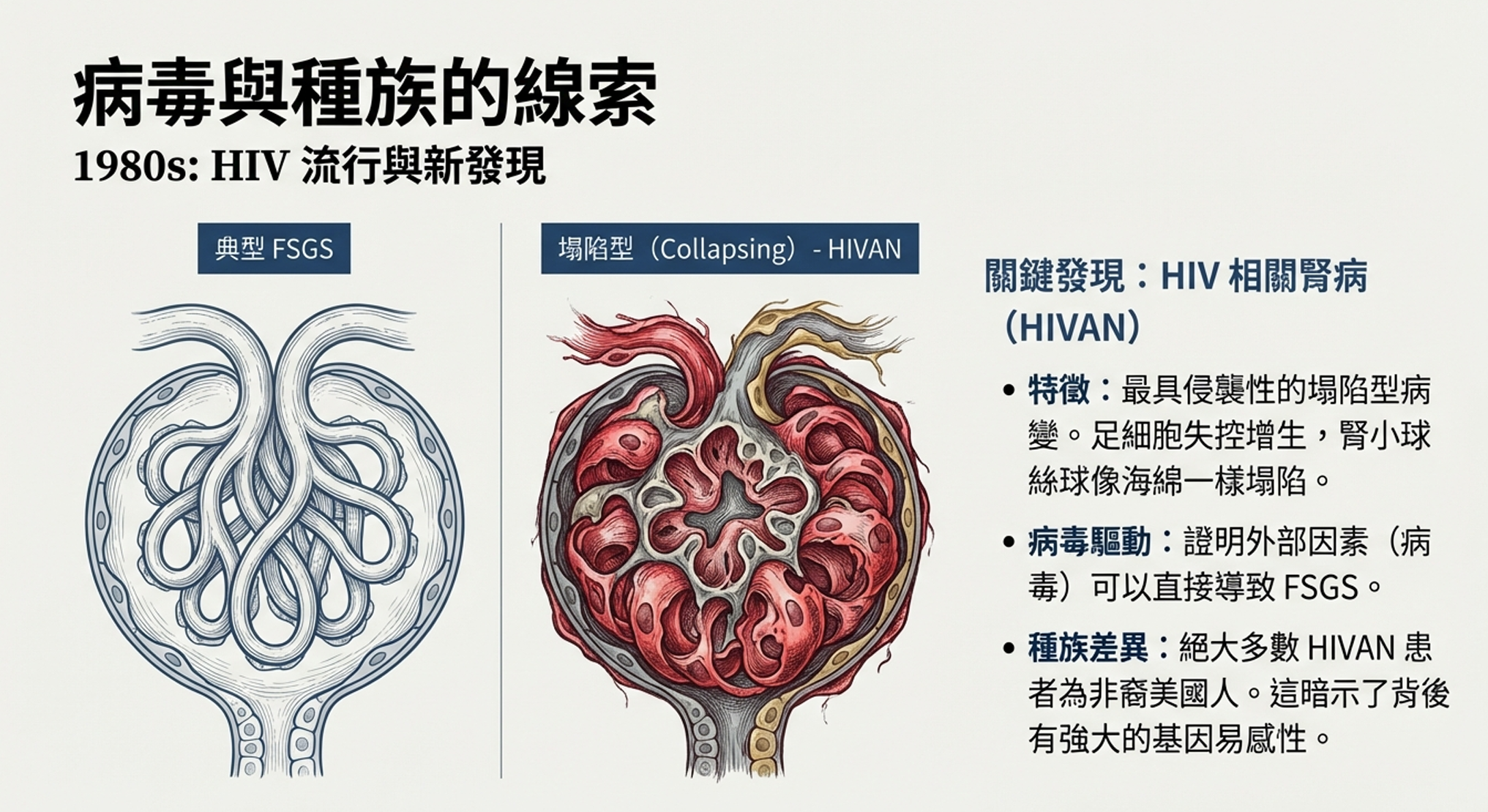
活檢顯示的是FSGS——但這不是普通的FSGS。
病理切片令人震驚。腎小球不只是部分硬化,它們在塌陷。整個腎小球絲球收縮成一團,像被擠壓過的海綿。更奇怪的是,那些本應附著在腎小球表面、扁平如鋪路石的足細胞,竟然增生、腫脹,填滿了整個尿腔。
「這看起來像是足細胞失控了,」病理科醫師Nicastri描述道。「它們不再是安靜的支撐細胞。它們在增殖,在改變形態,幾乎像是某種腫瘤性的變化。」
1984年3月,Rao的團隊在《新英格蘭醫學雜誌》發表了他們的發現。這篇論文不僅描述了一種新的疾病實體——後來被命名為HIV相關腎病(HIVAN)——更開創了一個重要概念:病毒可以直接導致FSGS。
這挑戰了「特發性」的標籤。FSGS不再只是原因不明的疾病,它可以是感染的後果。
更重要的是,Rao注意到一個奇怪的流行病學現象:幾乎所有HIVAN患者都是非裔美國人。即使控制了HIV感染率,黑人患者發展HIVAN的風險仍然遠高於白人。
「這裡面一定有基因的因素,」Rao在論文討論中寫道。但他不知道,這個觀察將在四分之一世紀後引發腎臟病學最重要的基因發現。
HIVAN的識別帶來了直接的臨床意義:這些患者需要的不是免疫抑制,而是抗反轉錄病毒治療。當高效抗反轉錄病毒療法(HAART)在1990年代後期問世時,HIVAN的發病率急劇下降。控制病毒,就能控制腎臟病。
但HIVAN留下的遺產遠不止於此。它確立了「塌陷型」作為FSGS最具侵襲性的病理變異型的概念。它暗示了非裔人群的獨特易感性。它證明了一個外部因素——病毒——可以驅動足細胞走向災難性的表型轉變。
這些線索將在未來的研究中匯聚,揭示一個關於人類演化、寄生蟲、和腎臟脆弱性的史詩故事。
第三章:給混亂命名(2000-2004)
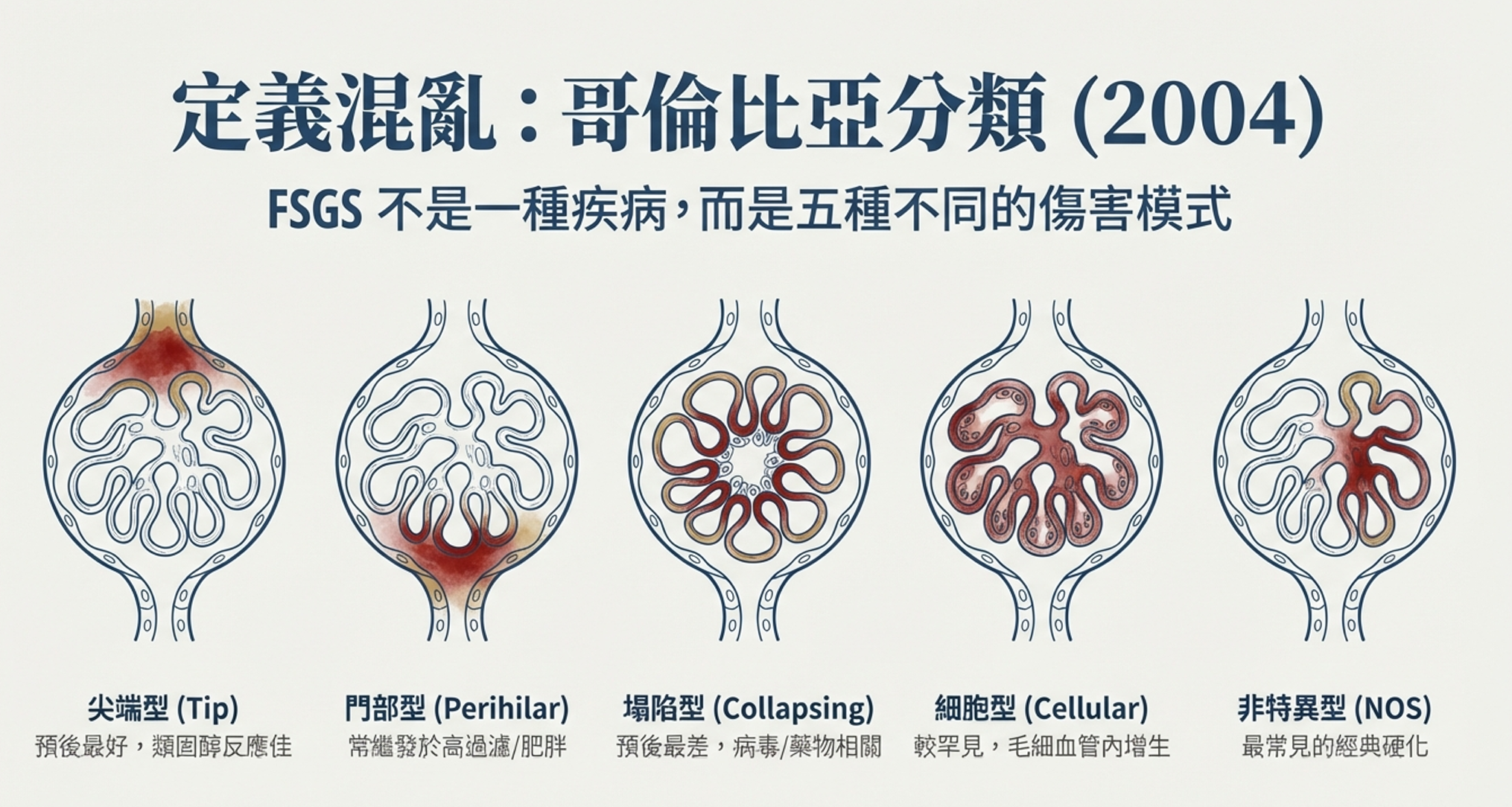
千禧年之交,FSGS的診斷陷入了混亂。
在全球各地的病理科實驗室裡,同一份活檢切片可能收到截然不同的報告。一位病理學家稱之為「FSGS」,另一位可能認為只是「微小病變伴局灶硬化」。有些硬化病變位於腎小管開口附近,有些位於血管進入處,有些則呈現整個腎小球的塌陷——它們都被籠統地歸類為「FSGS」,儘管臨床表現和預後可能天差地別。
臨床醫師們感到沮喪。「告訴我病人是FSGS毫無意義,」一位腎臟科醫師抱怨。「那就像告訴我病人『發燒』一樣。我需要知道是什麼類型的FSGS,才能預測他的預後,才能決定治療方案。」
這種混亂吸引了Vivette D'Agati的注意。
D'Agati是哥倫比亞大學的腎臟病理學家,以其敏銳的觀察力和系統性思維著稱。她已經花了二十年時間研究腎小球疾病,見過數千份FSGS活檢。在她看來,這些所謂「相同」的病變實際上講述著完全不同的故事。
「看這個,」她指著一張切片照片向她的同事們展示。「硬化病變正好位於近端腎小管的起點——我們稱為尖端的位置。這類患者通常發病急性,大量蛋白尿,但對類固醇反應很好。」
「現在看這個,」她換了一張照片。「硬化在血管極,靠近入球小動脈。你看到血管壁的玻璃樣變性了嗎?這是門部病變。這類患者通常有肥胖、高血壓、或者只有一個腎臟。他們的問題是血流動力學壓力,不是免疫因子。給他們用類固醇是浪費時間。」
「還有這個——」第三張照片更為駭人。腎小球完全塌陷,足細胞瘋狂增生。「塌陷型。HIV患者常見這種,預後最差。」
2000年11月4日至5日,一群志同道合的國際腎臟病理學家聚集在紐約哥倫比亞大學。D'Agati是這次會議的主要組織者。與會者來自美國、歐洲和日本,每個人都帶著自己的病例和困惑。
經過兩天激烈的討論和幻燈片檢視,一個共識開始浮現。他們決定將FSGS分為五種互斥的組織學變異型:
- 塌陷型——腎小球塌陷伴足細胞增生,最具侵襲性,快速進展至腎衰竭
- 尖端型——病變位於腎小管極,預後最好,類固醇反應率高
- 細胞型——毛細血管內增生明顯,罕見,可能是其他類型的早期階段
- 門部型——病變位於血管極伴玻璃樣變,常為繼發性
- 非特異型(NOS)——不符合上述任何類型的經典硬化,最常見
2004年,這個分類系統正式發表,被稱為「哥倫比亞分類」。它不是完美的——有些病例仍然難以歸類,有些變異型之間的界限模糊——但它提供了一種共同語言。
從此,當病理學家報告「塌陷型FSGS」時,臨床醫師就知道要準備壞消息:這個病人可能在兩年內需要透析。當報告是「尖端型」時,則可以更樂觀:有很大機會用類固醇達到緩解。
哥倫比亞分類的另一個重要貢獻是它的哲學轉變。它明確承認FSGS不是單一疾病,而是損傷模式——不同的原因可以導致相似的組織學外觀。這為後來的基因發現和機制分類鋪平了道路。
「我們不是在命名疾病,」D'Agati後來解釋。「我們是在描述足細胞受傷後的不同表現方式。真正的問題是:是什麼傷害了足細胞?」
這個問題的答案將從基因革命中浮現。
第二部:基因的密碼
第四章:芬蘭的詛咒(1998)
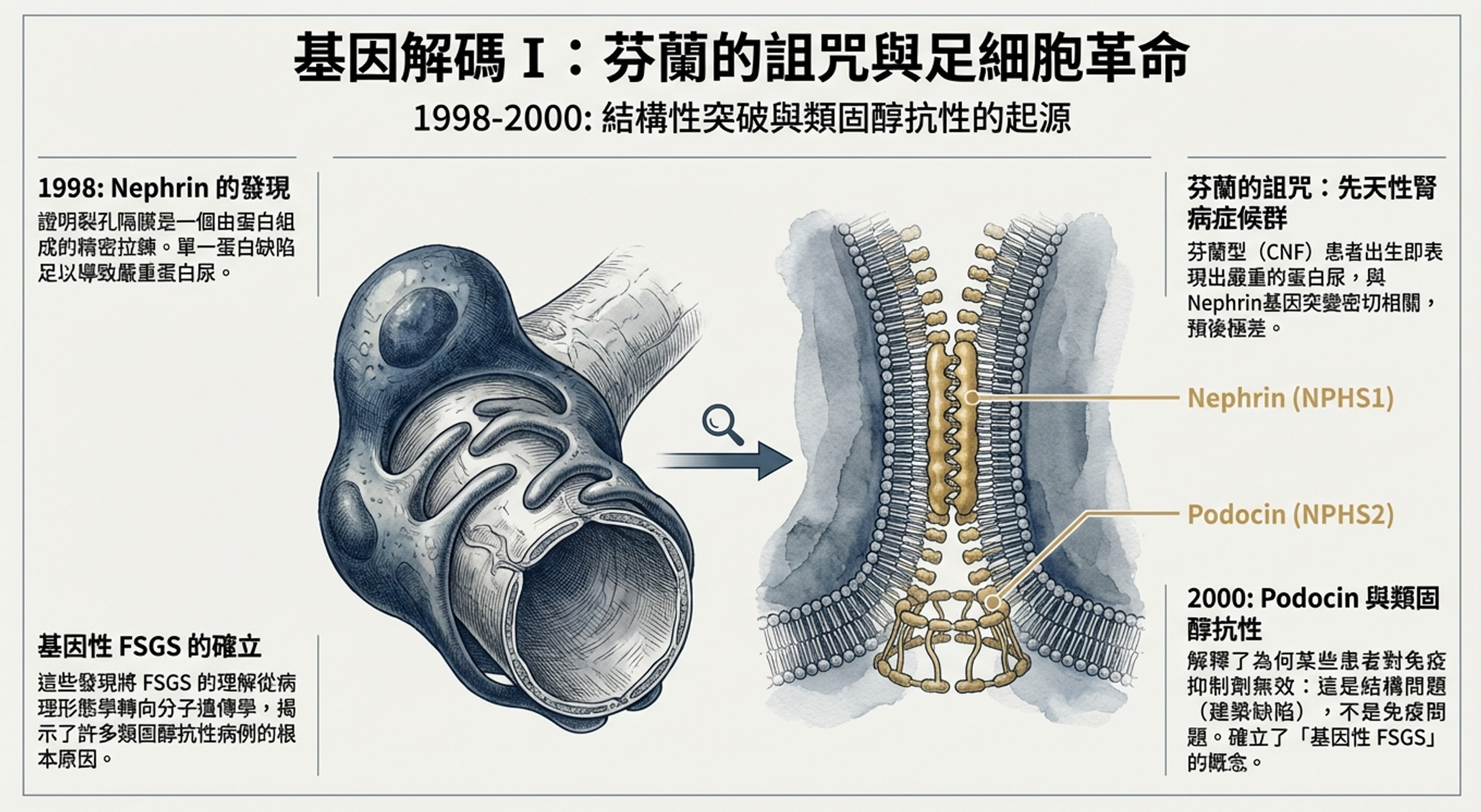
1990年代中期,芬蘭赫爾辛基大學的實驗室裡,Marjo Kestilä和她的團隊正在進行一場基因狩獵。
他們追蹤的是一種罕見但毀滅性的疾病:芬蘭型先天性腎病症候群。這種疾病幾乎只在芬蘭人群中出現,患病的嬰兒出生時就帶有腫脹的腹部和巨大的胎盤。他們從第一天起就失去大量蛋白質,如果不進行腎臟移植,很少能活過兩歲。
「這是芬蘭的詛咒,」一位當地醫師苦澀地說。在芬蘭,每八千名新生兒中就有一人罹患此病——這個發病率在世界其他地方幾乎聞所未聞。
芬蘭人群的獨特遺傳歷史——由少數創始者繁衍而來,經歷數千年的相對隔離——使得某些隱性疾病在這個民族中富集。這是悲劇,但對基因學家來說也是機會:這種「奠基者效應」使致病基因更容易被追蹤。
Kestilä的團隊使用位置克隆技術,這是一種在基因組中地毯式搜索致病突變的方法。經過數年的連鎖分析和精細定位,他們將目標縮小到染色體19上一個150kb的區域。
然後是艱苦的篩選工作:對這個區域的每個基因進行定序,尋找在患者和正常人之間不同的變異。
1998年的某一天,答案終於出現。
「我們找到了,」Kestilä宣布。致病基因編碼一種前所未知的蛋白質,他們將其命名為nephrin——源自希臘語「nephros」(腎臟)。
Nephrin是一種跨膜蛋白,屬於免疫球蛋白家族。它的表達高度特異——在整個人體中,只有腎小球的足細胞產生這種蛋白質。更精確地說,nephrin位於足細胞之間的裂孔隔膜——那層精巧的「拉鏈」結構,是腎小球過濾屏障的最後一道防線。
芬蘭患者中最常見的突變是「Fin-major」(121delCT),這是一個兩個核苷酸的缺失,導致移碼和蛋白質截短。沒有功能性nephrin,裂孔隔膜無法形成,蛋白質就像水通過破損的篩子一樣流入尿液。
這篇發表在《分子細胞》雜誌上的論文可能是腎小球生物學歷史上最重要的基礎科學發現。
它徹底改變了我們對腎臟過濾的理解。在此之前,教科書告訴學生,腎小球基底膜上的電荷屏障是防止蛋白質流失的主要機制。Nephrin的發現揭示了一個全新的世界:裂孔隔膜不只是被動的屏障,它是一個複雜的信號樞紐,由多種相互作用的蛋白質組成。
更重要的是,它證明了一個革命性概念:單一足細胞蛋白的缺陷就足以導致災難性的蛋白尿。這為理解FSGS打開了全新的大門——如果基因突變能導致先天性腎病症候群,那麼獲得性FSGS是否也涉及相同蛋白質的功能障礙?
答案很快就會揭曉。
第五章:第二把鑰匙(2000)
Nephrin的發現像投入池塘的石子,激起一圈圈漣漪。全世界的研究者開始搜尋其他可能導致遺傳性腎病症候群的基因。
在法國巴黎,Corinne Antignac的團隊已經在追蹤另一種遺傳性腎臟病:常染色體隱性類固醇抗性腎病症候群。這些患者不像芬蘭型那樣出生即發病,而是在兒童期出現腎病症候群。他們的活檢顯示FSGS,而非微小病變。最關鍵的是:無論使用多少類固醇,他們的蛋白尿都不消退。
家族分析顯示這是隱性遺傳——患者的父母是健康的攜帶者,只有繼承兩個突變拷貝的孩子才會發病。
2000年4月,Antignac團隊在《自然遺傳學》發表了他們的發現:致病基因是NPHS2,編碼一種新蛋白質podocin。
Podocin定位於足細胞的裂孔隔膜,就在nephrin旁邊。它是一種錨定蛋白,幫助將nephrin固定在正確的位置,並調節其信號功能。當podocin突變時,整個裂孔隔膜複合物變得不穩定。
這項發現的臨床意義是革命性的。
「這解釋了為什麼有些孩子對類固醇完全無反應,」Antignac說。「他們的問題不是免疫失調。他們的問題是建築缺陷——腎臟過濾器的結構本身就有缺陷。對這些患者使用免疫抑制劑,就像試圖用抗生素治療斷腿一樣荒謬。」
更實際的發現來自對特定突變的分析。研究團隊發現一個常見的變異——R229Q——在歐洲人群中的等位基因頻率約為3.6%。這個變異單獨不會致病,但當它與另一等位基因上的嚴重突變配對時,會導致成人發病型FSGS。
這解釋了一個長期困惑臨床醫師的現象:為什麼有些FSGS患者三十多歲才發病,而且家族中似乎沒有其他人受影響?答案是複合雜合性——患者從父母分別繼承了不同的突變,而父母作為攜帶者都是健康的。
Podocin的發現確立了「基因性FSGS」作為一個獨特的類別。對這些患者,基因檢測現在取代了漫長而有毒的免疫抑制試驗。更重要的是,它為移植提供了好消息:基因突變導致的FSGS在移植後幾乎不會復發,因為問題源於患者自身的足細胞基因,而移植腎帶有健康的基因。
兩項發現加在一起,nephrin和podocin定義了一個新的研究領域:足細胞生物學。這些曾經被忽視的細胞——在教科書中只被簡單描述為「覆蓋腎小球毛細血管的上皮細胞」——現在被認識到是腎小球過濾的核心組件,是一個複雜分子機器的載體。
科學家們開始繪製「裂孔隔膜蛋白質組」——這個精巧結構中所有相互作用的分子。他們發現nephrin和podocin只是冰山一角。CD2AP、FAT1、NEPH1、α-actinin-4、TRPC6……一個又一個蛋白質被發現參與這個超分子複合物。每一個都是潛在的疾病基因。
FSGS不再是一個「垃圾桶診斷」。它正在被解構成數十種不同的分子疾病。
第六章:骨架的秘密(2000-2010)
如果說nephrin和podocin揭示了裂孔隔膜的重要性,接下來的發現則將目光轉向足細胞的另一個關鍵結構:細胞骨架。
2000年,就在podocin發現的同一年,波士頓的Jonathan Kaplan團隊報告了一個意想不到的發現。他們研究的是一種罕見的家族性FSGS——不是隱性遺傳,而是顯性遺傳。這意味著只需要一個突變拷貝就足以致病,疾病會代代相傳。
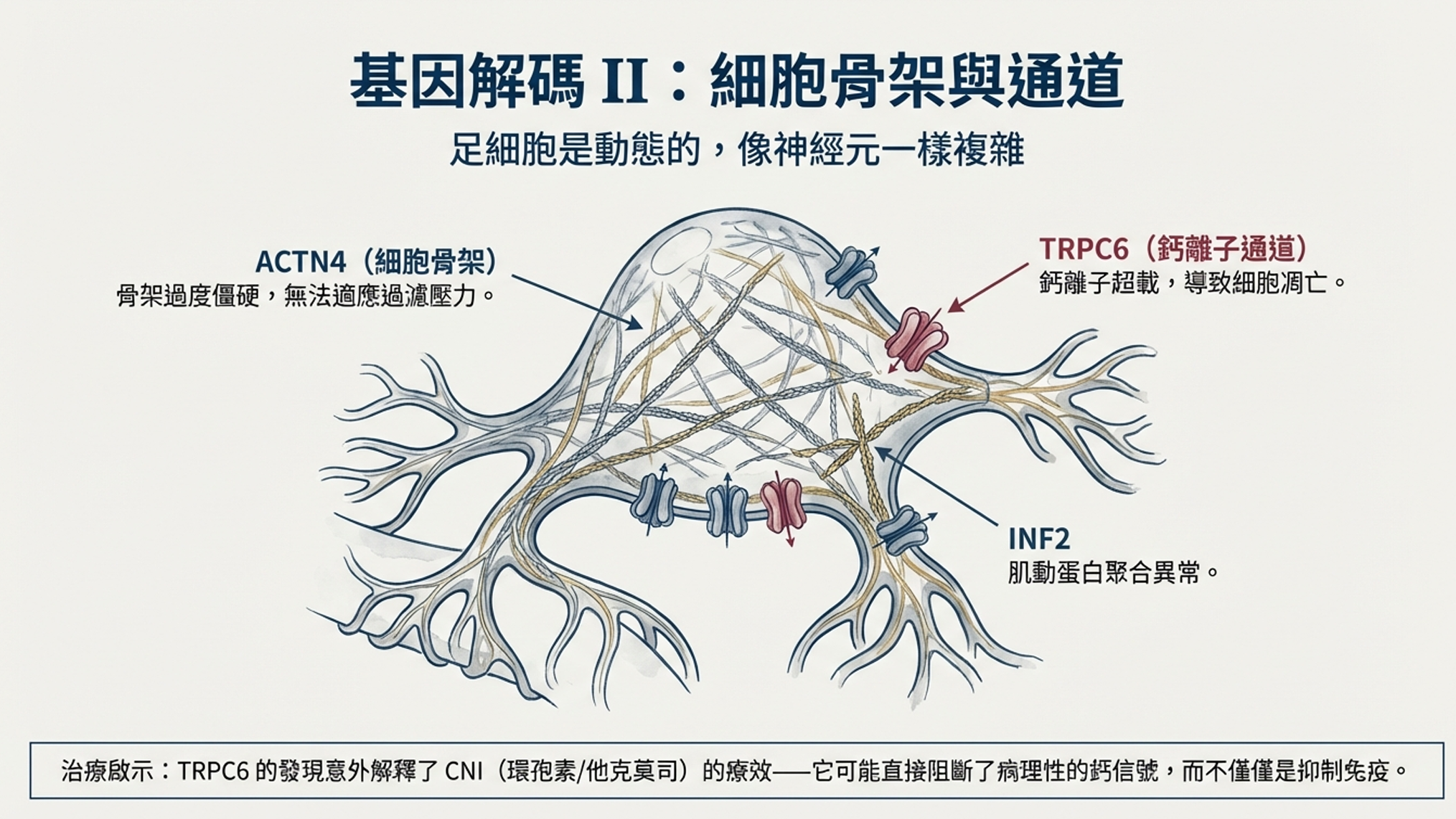
致病基因是ACTN4,編碼α-actinin-4——一種肌動蛋白絲交聯蛋白。
這與nephrin和podocin的故事截然不同。Nephrin和podocin位於細胞表面的裂孔隔膜;α-actinin-4則在細胞內部,是構成足細胞「骨架」的核心成分。
更有趣的是突變的機制。大多數致病基因突變導致功能喪失——蛋白質無法正常工作。但ACTN4突變是功能獲得。突變型α-actinin-4與肌動蛋白絲的結合比野生型更強,而不是更弱。
「這聽起來應該是好事,」Kaplan解釋,「但實際上是災難。足細胞需要動態地重塑其細胞骨架,以應對腎小球過濾時的血流動力學壓力。如果α-actinin-4與肌動蛋白結合得太緊,細胞骨架就變得僵硬。這些突變蛋白還會在細胞內形成聚集體——類似於神經退行性疾病中看到的蛋白聚集。」
這個發現將足細胞描繪為一種動態的、收縮性的細胞——更像肌肉細胞或神經元,而非靜態的上皮細胞。它必須不斷調整自己的形狀,緊緊附著在腎小球基底膜上,同時承受過濾壓力。
五年後,另一項發現進一步豐富了這個圖景。
2005年,Martin Winn在《科學》雜誌報告了一個大型紐西蘭家族的遺傳性FSGS。致病基因是TRPC6——一種鈣離子通道。
TRPC6位於足細胞的細胞膜上,允許鈣離子流入細胞。當它突變時,通道變得過度活躍,過多的鈣離子湧入足細胞。
「鈣是細胞內的二級信使,」Winn解釋。「它控制著無數的細胞過程。鈣超載會激活鈣調神經磷酸酶-NFAT通路,改變基因表達,最終導致足細胞凋亡。」
這個發現為一種常用藥物提供了意想不到的理論支持:鈣調神經磷酸酶抑制劑(CNIs),如環孢素和他克莫司。
這些藥物最初被開發用於器官移植的免疫抑制,後來被發現對類固醇抗性FSGS有效。傳統解釋是它們抑制T細胞,減少了Shalhoub假設的「通透因子」產生。但TRPC6的發現暗示了另一種可能性:CNIs可能直接保護足細胞,通過阻斷病理性鈣信號。
2010年,最後一塊重要的拼圖落入位置。Elizabeth Brown的團隊發現INF2突變是家族性FSGS最常見的原因之一。
INF2是一種formin蛋白——調節肌動蛋白聚合和解聚的酶。有趣的是,INF2突變不僅導致腎臟病,還與Charcot-Marie-Tooth病(一種周邊神經病變)相關。一些患者同時患有兩種疾病。
「足細胞和神經元有很多共同點,」Brown指出。「它們都是高度特化的、有絲分裂後的細胞,依賴複雜的細胞骨架來維持其獨特的形態。足細胞的足突就像神經元的軸突和樹突一樣,需要精確的細胞骨架調節。」
到2010年,FSGS的基因版圖已經相當壯觀。研究者識別出數十個致病基因,它們大致可分為幾類:
- 裂孔隔膜蛋白(nephrin, podocin, CD2AP)
- 細胞骨架調節蛋白(ACTN4, INF2, MYO1E)
- 離子通道(TRPC6)
- 轉錄因子(WT1, LMX1B)
- 線粒體蛋白(COQ2, COQ6)
每一個新發現的基因都講述著足細胞如何維持其功能的故事,也揭示了這些精密機器可能失敗的方式。
但最大的發現還在前方——它將來自對一個古老演化謎題的解答。
第七章:非洲的遺產(2010)
2010年夏天,一篇發表在《科學》雜誌上的論文震撼了整個腎臟病學界。
數十年來,流行病學家們困惑於一個殘酷的事實:非裔美國人發展末期腎病(ESKD)的風險是歐裔美國人的四倍。這種差異無法完全用社會經濟因素、醫療可近性、或高血壓控制差異來解釋。
傳統智慧將許多這些病例歸類為「高血壓性腎硬化」——高血壓導致的腎臟損傷。但有些腎臟科醫師懷疑這個標籤過於簡化。這些患者的腎臟活檢常常顯示FSGS,而非典型的高血壓性改變。是否存在某種特定的基因變異使非裔人群的腎臟更加脆弱?
2008年,研究者首先將信號定位到MYH9基因——編碼非肌肉肌球蛋白重鏈IIA,這是細胞骨架的另一個組件。但後續研究發現這個關聯並不完美,統計信號似乎來自鄰近區域。
Giulio Genovese和David Friedman在Martin Pollak的實驗室決定深入挖掘。他們使用更精細的基因定位技術,結合混血譜系分析(利用非裔美國人基因組中混合的非洲和歐洲血統來追蹤疾病基因)。
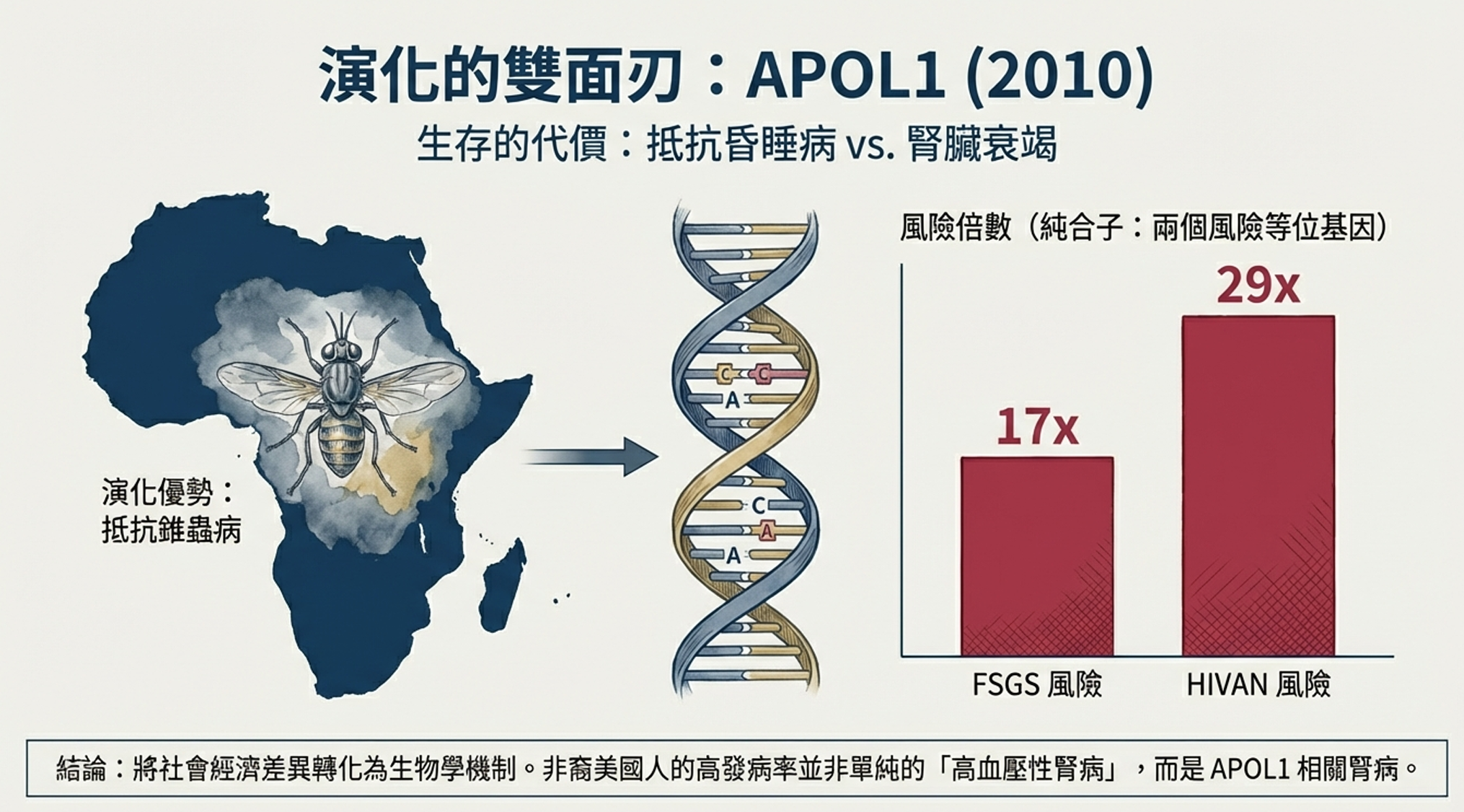
答案指向MYH9旁邊的一個基因:APOL1。
APOL1編碼載脂蛋白L1,這是一種在血液中循環的蛋白質。令人驚訝的是,它的主要已知功能與腎臟毫無關係——它是人類先天免疫系統對抗非洲錐蟲的武器。
故事要追溯到數千年前的非洲大陸。
布氏錐蟲是一種寄生蟲,由采采蠅傳播,導致非洲昏睡病——一種不治療必死的疾病。人類是少數幾種對普通布氏錐蟲具有天然抵抗力的哺乳動物,這要歸功於血液中的「溶錐蟲因子」,其中APOL1是關鍵成分。APOL1可以穿透錐蟲的細胞膜,殺死這些寄生蟲。
但有一種特別危險的亞種——羅德西亞布氏錐蟲——演化出了逃避機制,能夠抵抗正常的APOL1。它在東非造成致命的昏睡病。
大約一萬年前,在人類與這種致命寄生蟲的軍備競賽中,兩個APOL1變異在非洲人群中出現並迅速擴散:G1(兩個錯義突變的組合)和G2(一個六核苷酸缺失)。這些變異產生的改變型APOL1蛋白能夠殺死羅德西亞錐蟲,提供對昏睡病的保護。
這是雜合子優勢的經典例子——與鐮刀細胞性狀保護瘧疾類似。攜帶一個G1或G2拷貝的人對昏睡病具有抵抗力,同時腎臟相對健康。
問題出在純合子或複合雜合子——那些繼承了兩個風險等位基因(G1/G1, G2/G2, 或G1/G2)的人。對他們來說,保護變成了詛咒。
Genovese團隊的數據令人震驚:
- 擁有兩個APOL1風險等位基因的人,發展FSGS的風險是普通人的17倍
- 發展HIV相關腎病的風險是29倍
- 約13%的非裔美國人攜帶兩個風險等位基因
「這解釋了一切,」Friedman後來回憶。「高血壓性腎病、HIVAN、非糖尿病性ESKD——所有這些在非裔美國人中過度代表的疾病,現在都可以用APOL1來理解。這不是多種不同疾病的巧合,而是同一個基因易感性的不同表現。」
APOL1的發現改變了我們對種族健康差異的理解。數十年來,腎臟病學界傾向於從社會決定因素的角度解釋這些差異——貧困、歧視、醫療不平等。這些因素當然重要,但APOL1揭示了一個不可忽視的生物學層面:演化歷史留在基因中的印記。
它還帶來了直接的臨床意義:
- 診斷重新分類:許多之前被標記為「高血壓性腎病」的非裔患者,實際上應該診斷為「APOL1相關腎病」
- 移植考量:非裔美國人活體腎臟捐贈者如果攜帶兩個APOL1風險等位基因,他們自己發展腎病的風險顯著增加,需要在捐贈前進行遺傳諮詢
- 治療開發:APOL1成為藥物開發的靶標,多家製藥公司開始開發APOL1抑制劑
- COVID-19的啟示:2020年新冠大流行期間,醫師們觀察到非裔患者中不成比例的腎臟損傷。APOL1框架幫助解釋了這一現象——病毒感染可能作為「二次打擊」,在基因易感者中觸發腎臟病
APOL1的故事是科學如何揭示人類歷史的典範。一種在非洲草原上提供生存優勢的基因變異,隨著跨大西洋奴隸貿易被帶到新世界,在那裡——遠離采采蠅和昏睡病的威脅——它成為數百萬人腎臟脆弱性的來源。
這是達爾文演化的陰暗面:有利的變異可能在環境改變後成為負擔。但同時,這也是希望的來源:理解機制意味著可以開發治療方法。APOL1抑制劑正在臨床試驗中,如果成功,將為非裔患者帶來第一個針對其特定遺傳風險的精準藥物。
第三部:尋找幽靈
第八章:血液中的信號(2011)
即使基因革命如火如荼地展開,腎臟病學家們並沒有忘記Shalhoub四十年前提出的問題:是否存在一種循環因子,在原發性、非基因性FSGS中驅動蛋白尿?
最強有力的臨床證據來自腎臟移植。
想像這個場景:一名特發性FSGS患者的腎臟徹底衰竭,她接受了腎臟移植。手術順利,新腎臟來自健康的捐贈者。然而,就在手術後幾小時——甚至在手術室還沒關閉的時候——新腎臟開始大量漏出蛋白質。
這不可能是免疫排斥——那需要數天到數週才會發生。這不可能是捐贈者腎臟的問題——捐贈者是健康的,而且這種現象在受者的第一次移植時就可能出現,而捐贈者腎臟在其他受者體內可能完全正常。
唯一的解釋是:患者的血液中存在某種物質——某種循環因子——在接觸到新腎臟的那一刻就開始攻擊它。
這種「即刻復發」現象在約三分之一的特發性FSGS患者中發生。如果用血漿置換清除血液中的蛋白質,蛋白尿通常會減輕——直到停止血漿置換,蛋白尿再次回來。
幾十年來,研究者嘗試識別這個因子。他們將FSGS患者的血清加入體外培養的腎小球或足細胞,觀察通透性變化。他們用各種生化方法分離和純化血漿成分,尋找「活性組分」。
1996年,Virginia Savin的團隊報告他們可以用FSGS患者血清增加離體大鼠腎小球的白蛋白通透性。這個「通透活性」可以被血漿置換降低,與臨床觀察一致。但他們無法識別確切的分子。
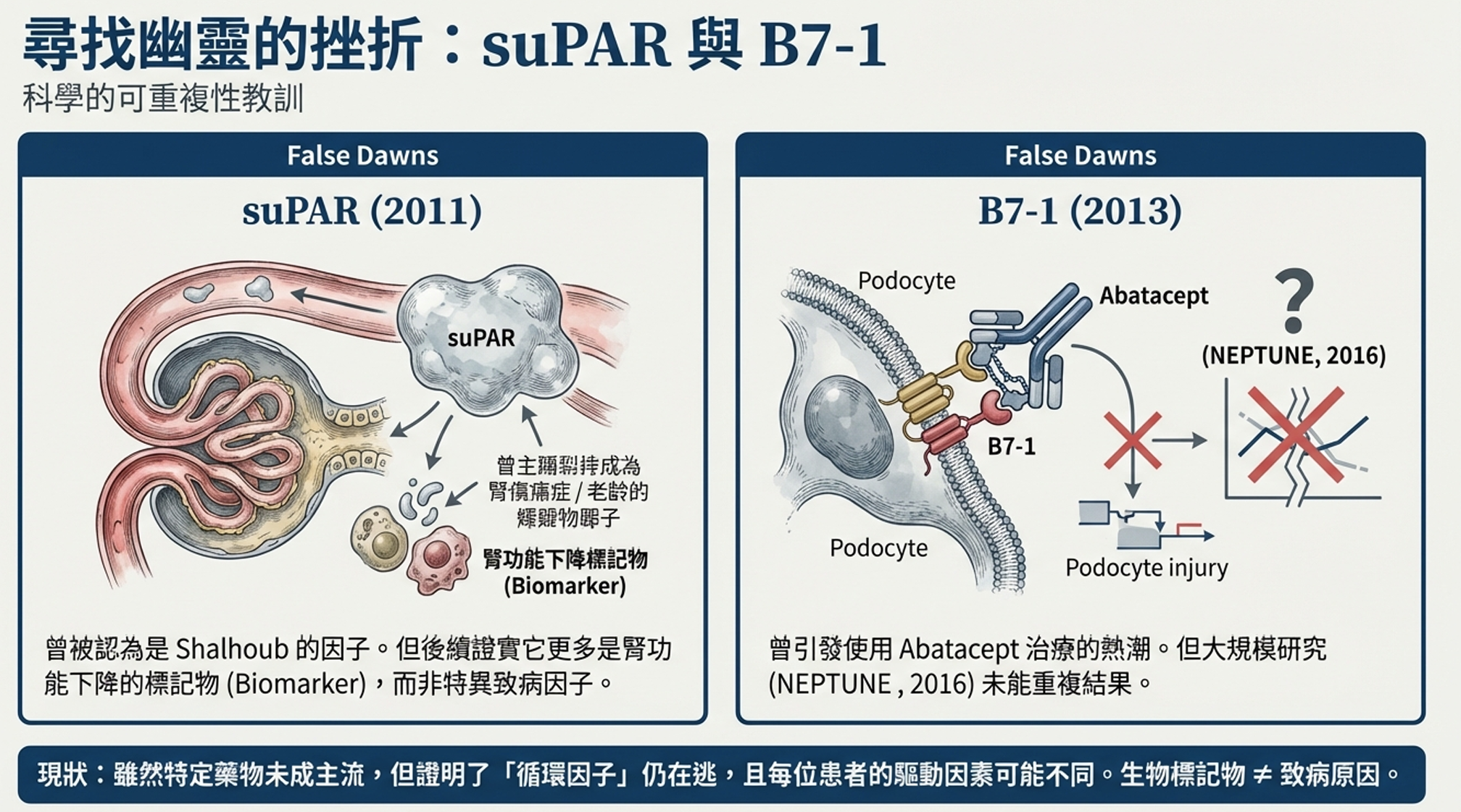
2011年,芝加哥的Jochen Reiser團隊宣布了一個激動人心的發現:他們聲稱找到了那個幽靈。
它是suPAR——可溶性尿激酶纖溶酶原激活物受體。
Reiser的論文發表在《自然醫學》,證據鏈看起來令人信服:
- 原發性FSGS患者的血清suPAR水平高於其他腎臟病患者
- suPAR可以激活足細胞上的β3整合素,導致足突融合
- 將suPAR注射到小鼠體內可以誘導蛋白尿和類FSGS病變
「我們可能終於找到了Shalhoub預言的那個因子,」Reiser說。論文暗示suPAR可能成為診斷原發性FSGS的生物標記物,以及治療的靶標。
興奮迅速蔓延。世界各地的腎臟科醫師開始測量患者的suPAR水平。製藥公司開始探索阻斷suPAR或整合素的策略。
但科學是殘酷的——它要求可重複性。
2015年,由Jonathan Spinale領導的團隊使用NEPTUNE隊列——一個精心設計、特徵明確的腎小球疾病患者數據庫——嘗試驗證suPAR假說。
結果令人失望。
「suPAR水平確實在FSGS患者中升高,」Spinale報告,「但這可以完全用腎功能來解釋。suPAR由腎臟清除。當腎功能下降時,suPAR積累。我們發現的只是腎功能不全的標記物,而非FSGS的特異性因子。」
他們的小鼠實驗也未能重現Reiser團隊的結果。即使維持高濃度suPAR數週,動物也沒有發展出腎臟病。
suPAR假說並未被完全否定——科學辯論仍在繼續——但它的最初光芒已經褪色。suPAR可能是腎臟病進展的標記物,但將其稱為FSGS的「致病因子」過於武斷。
這場爭議留下了重要的教訓:在罕見病研究中,獨立驗證至關重要。轟動性的發現必須經受嚴格審視。生物標記物的「升高」不等於「因果」。
幽靈因子仍然在逃。
第九章:足細胞上的旗幟(2013-2016)
2013年,另一個激動人心的假說出現——這一次,它承諾的不僅是診斷標記物,還有現成的治療方法。
Peter Mundel是哈佛大學的足細胞專家,他的團隊報告在蛋白尿患者的腎臟活檢中發現一種令人驚訝的分子:B7-1(也稱為CD80)。
B7-1通常在免疫細胞上表達——特別是抗原呈遞細胞。它是T細胞激活所需的「共刺激」分子。它不應該出現在足細胞上。
但Mundel的團隊聲稱,在某些腎臟病患者中,足細胞異常地表達B7-1。他們假設這種異位表達破壞了足細胞的細胞骨架穩定性,導致蛋白尿。
如果這是真的,治療方法就在眼前:Abatacept(CTLA-4-Ig)是一種已經批准用於類風濕性關節炎的藥物,它專門阻斷B7-1。
Mundel團隊報告了五名患者的「慈悲使用」經驗。這些患者都是治療難治性病例——有些是移植後復發、對其他所有治療無反應的絕望案例。在確認他們的活檢標本B7-1染色陽性後,給予Abatacept治療。
結果看起來驚人:蛋白尿「戲劇性、快速地」緩解。
這篇論文發表在《新英格蘭醫學雜誌》——醫學界最具影響力的期刊之一。它被譽為FSGS「精準醫學」的曙光。如果可以通過活檢識別B7-1陽性患者,然後用現成的藥物治療,這將是一個突破。
臨床界迅速行動。世界各地的腎臟科醫師開始對活檢進行B7-1染色,對陽性患者使用Abatacept。病例報告和小型系列陸續發表,描述了成功的案例。
但問題開始浮現。
首先是可重複性問題。多個獨立的病理學團隊嘗試在FSGS活檢中檢測足細胞B7-1,使用各種抗體和方案,結果一無所獲。義大利的Benigni團隊、美國的Alachkar團隊都報告了陰性結果。他們質疑原始研究中的「陽性染色」是否是技術偽影。
「我們用了五種不同的抗體,各種固定和染色條件,」一位病理學家說。「我們看不到任何B7-1信號。」
然後是臨床試驗的失敗。Emory大學的Larry Greenbaum領導了一項前瞻性試驗(NCT02592798),評估Abatacept在FSGS中的療效。結果與最初的病例報告截然不同——Abatacept並不比對照組更有效,而且出現了嚴重不良事件。
B7-1假說的倒塌是一個警示故事:
- 小型、非對照的病例系列可能產生虛假的陽性結果
- 「戲劇性緩解」可能是自然病程、安慰劑效應、或其他同時治療的結果
- 免疫組織化學是一種敏感的技術,容易產生假陽性
- 在改變臨床實踐之前,需要嚴格的驗證和隨機對照試驗
2021年的KDIGO指南明確建議不要常規使用Abatacept治療FSGS。曾經被譽為突破的治療方法,現在被認為是未經證實的。
Shalhoub的幽靈因子仍未被捕獲。suPAR和B7-1都是它的偽裝,而非真身。
也許,就像量子物理學中的不確定性原理,這個因子的本質就是難以捉摸。也許它不是單一分子,而是多種因子的組合。也許它在不同患者中不同。也許它根本不存在,「原發性」FSGS實際上是一組尚未識別的基因疾病。
無論如何,搜索仍在繼續。每一次失敗都縮小了可能性空間,使我們更接近真相——無論那個真相是什麼。
第四部:治療的藝術
第十章:類固醇的困境(1974-1999)
讓我們暫時離開分子和基因的世界,回到臨床現實。
一名35歲的女性,腿部水腫嚴重,尿液泡沫如啤酒。腎臟活檢顯示FSGS。她的醫師面臨著一個古老的問題:怎麼治療她?
1970年代至1990年代,治療FSGS的選擇極為有限。基本策略是:
第一步:高劑量類固醇
Prednisone,每天1毫克/公斤體重,持續數月。這是一種強效的免疫抑制劑,對微小病變腎病非常有效。在FSGS中,類固醇的緩解率較低,但仍有相當一部分患者會有反應。
問題是副作用。長期高劑量類固醇導致滿月臉、水牛肩、骨質疏鬆、白內障、糖尿病、感染風險增加、情緒不穩、失眠……清單幾乎無窮無盡。患者常常面臨一個殘酷的選擇:忍受疾病,還是忍受治療?
第二步:如果類固醇失敗呢?
在1999年之前,答案基本是:不知道。
有些醫師嘗試環磷醯胺(cytoxan)——一種化療藥物,也是強效免疫抑制劑。它可能有效,但帶有嚴重毒性,包括不孕、膀胱癌風險、和骨髓抑制。
有些醫師嘗試環孢素(cyclosporine)——一種來自真菌的免疫抑制劑,最初開發用於器官移植。有病例報告顯示它可能有效,但沒有嚴格的試驗數據。
更多的醫師只能告訴患者:「我們已經盡力了。」然後眼睜睜看著他們的腎功能慢慢惡化,最終走向透析。
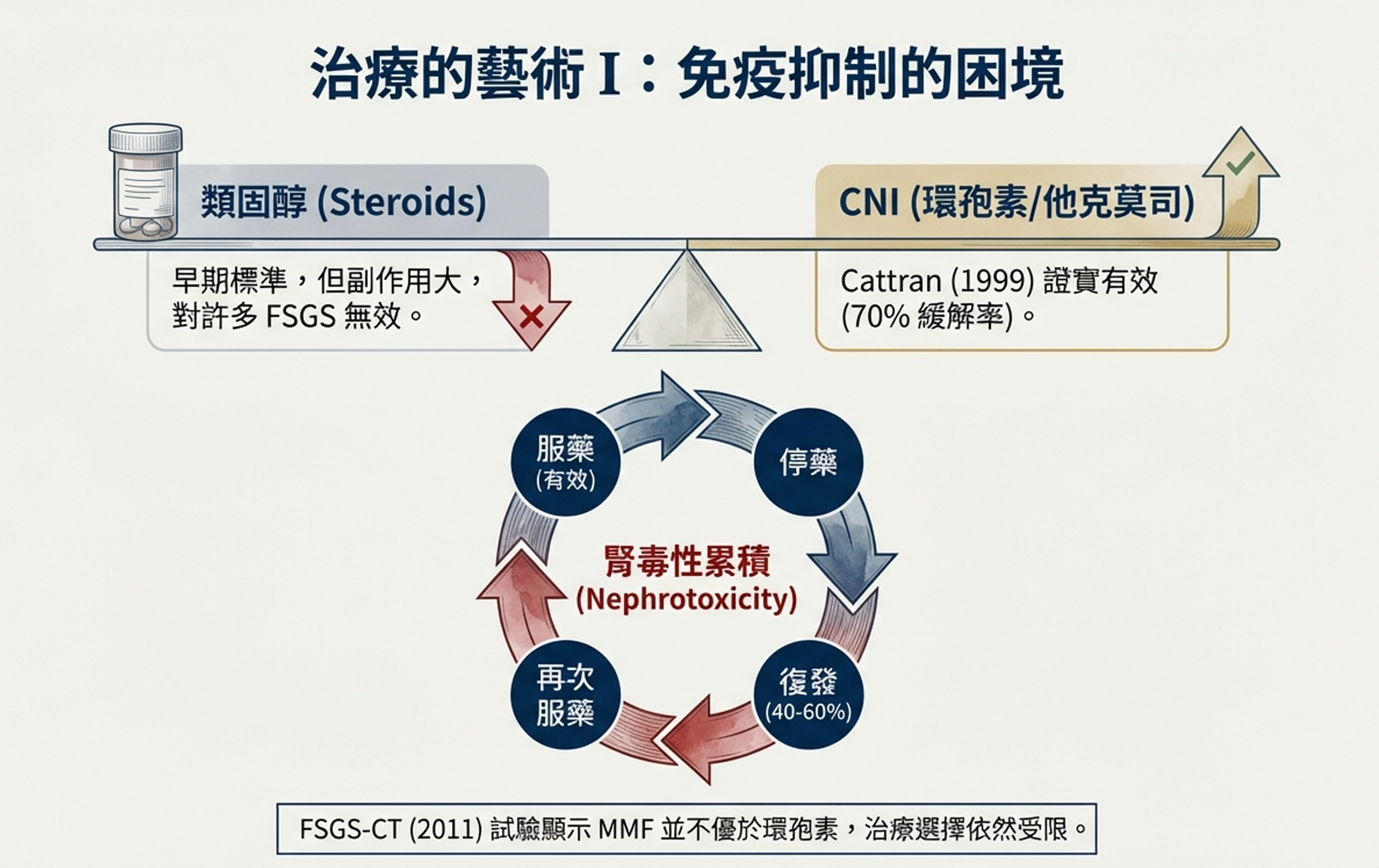
1999年,Daniel Cattran改變了這一切。
Cattran是加拿大多倫多大學的腎臟科醫師,他組織了北美腎病症候群研究組進行一項決定性的隨機對照試驗:環孢素對比安慰劑,在類固醇抗性FSGS患者中。
結果發表在《腎臟國際》(Kidney International)雜誌上:
- 環孢素組:70%的患者達到部分或完全緩解
- 安慰劑組:僅4%
這是壓倒性的證據。環孢素有效。
從此,鈣調神經磷酸酶抑制劑(CNIs)——環孢素和後來的他克莫司——成為類固醇抗性FSGS的標準二線治療。這寫入了指南,改變了全球的臨床實踐。
但Cattran的論文也揭示了一個令人沮喪的事實:即使用環孢素達到緩解,停藥後復發率高達40-60%。許多患者需要長期維持CNI治療,而CNI本身具有腎毒性——長期使用會損傷腎臟。
「這是一個困境,」Cattran承認。「我們用一種腎毒性藥物來治療腎臟病。我們必須在疾病的傷害和治療的傷害之間尋找平衡。」
第十一章:大試驗的教訓(2011)
Cattran的試驗確立了CNI的地位,但研究者們想知道:是否有更好的選擇?
黴酚酸酯(Mycophenolate Mofetil, MMF)是一種較新的免疫抑制劑,作用機制與CNI不同,而且沒有腎毒性。它被廣泛用於器官移植和系統性紅斑狼瘡。如果MMF在FSGS中同樣有效,它可能取代CNI成為更安全的選擇。
2003年,美國國立衛生研究院(NIH)決定資助一項大規模、多中心隨機試驗,直接比較CNI和MMF在兒童和青年類固醇抗性FSGS中的療效。這被稱為FSGS臨床試驗(FSGS-CT)。
Debbie Gipson是這項試驗的主要研究者之一。歷時數年,他們招募了138名患者——這是當時FSGS有史以來最大的隨機試驗。患者被隨機分配到接受12個月的環孢素治療,或口服脈衝地塞米松加黴酚酸酯的組合。
所有人都期待著結果能給出明確答案。
2011年,結果發表在《腎臟國際》:
「試驗未能顯示MMF/地塞米松優於環孢素」
緩解率:
- 環孢素組:33名患者達到部分或完全緩解
- MMF/地塞米松組:22名患者達到緩解
差異無統計學意義。而且,兩組的緩解率都令人失望——不到一半的患者對任何治療有反應。
更令人憂慮的是,在整個78週的研究期間,每組各有7-8名患者死亡或發展為腎衰竭。
FSGS-CT是一個「陰性」試驗,但它的影響深遠:
- 確認治療的困難:即使是積極的免疫抑制,多數類固醇抗性患者仍然無法達到緩解。FSGS是一種頑固的疾病。
- CNI仍是標準:既然MMF沒有顯示出優越性,環孢素(或他克莫司)仍然是二線治療的首選。
- 暗示異質性:回顧性分析發現,隊列中約30%的患者可能攜帶致病基因突變。這些基因性FSGS患者對免疫抑制無反應是可預期的。如果能事先排除這些患者,「真正的」免疫性FSGS可能會顯示更好的治療反應。
- 呼籲基因檢測:FSGS-CT強調了在入組前進行基因篩查的重要性。未來的試驗需要排除基因性病例,以更準確地評估免疫抑制的療效。
第十二章:Meyrier的智慧(2009)
在治療學的混戰中,有時需要一位智者來釐清思路。
Alain Meyrier是法國的資深腎臟科醫師,他花了數十年時間研究腎小球疾病。2009年,他發表了一篇影響深遠的綜述,標題平實:「FSGS治療選擇的更新」。但其中包含的哲學轉變比任何新藥物試驗都更重要。
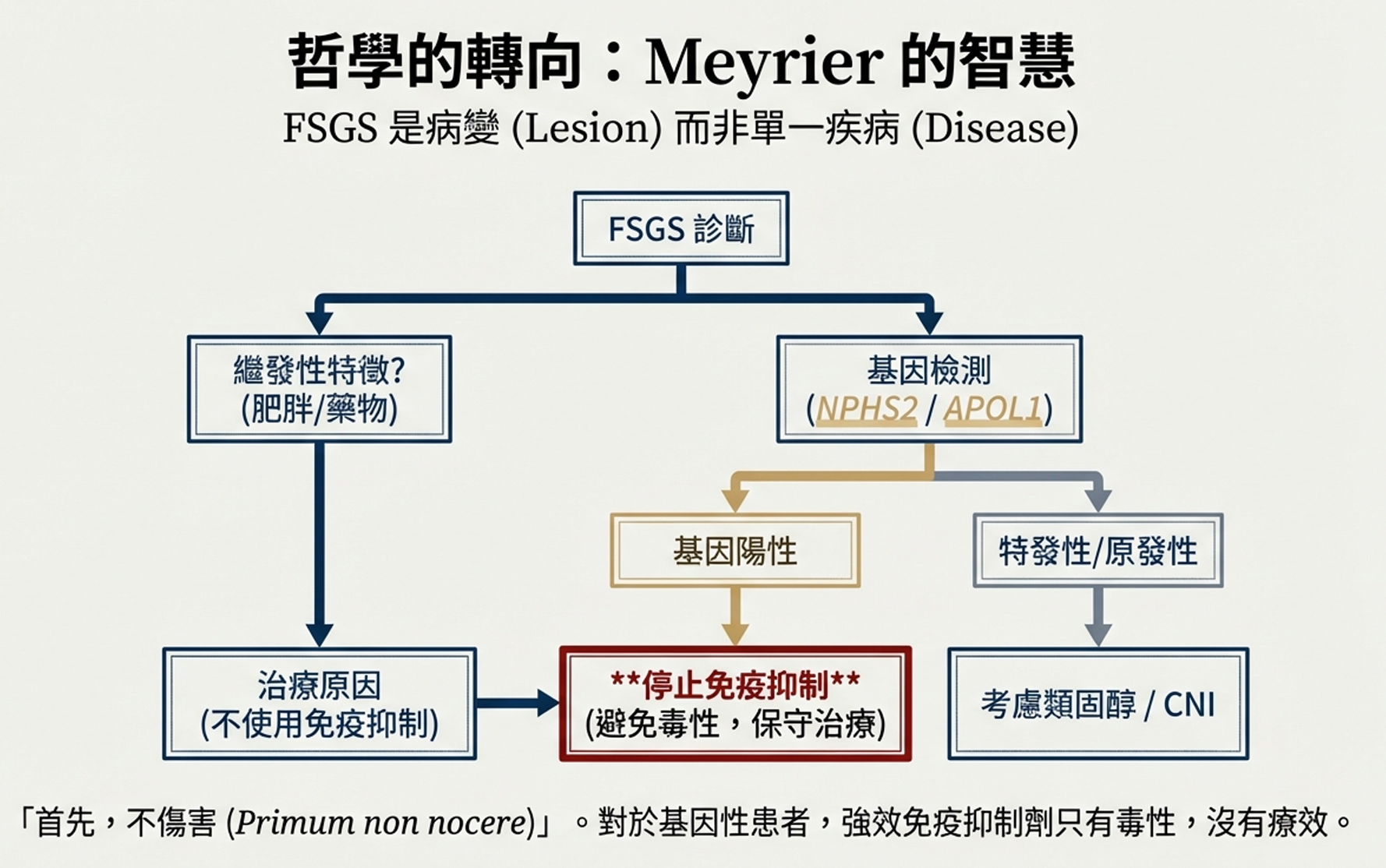
Meyrier的核心論點是:FSGS是一種病變,不是一種疾病。
「我們不能用單一的治療算法對待所有FSGS患者,」他寫道。「一個因肥胖和高過濾導致的門部型FSGS患者,和一個因循環因子導致的塌陷型患者,是完全不同的疾病。用相同的類固醇和CNI方案治療他們,不是精準醫學,而是無差別轟炸。」
他提出了一個臨床決策框架:
步驟1:確認診斷
↓
步驟2:尋找繼發原因(肥胖、病毒、藥物、適應性反應)
↓
如果找到 → 針對原因治療,不需要免疫抑制
↓
如果未找到 → 考慮原發性/特發性
↓
步驟3:嘗試類固醇
↓
如果反應 → 維持緩解
↓
如果抗性 → 步驟4
↓
步驟4:基因檢測
↓
如果發現致病突變 → 停止免疫抑制!專注保守治療
↓
如果無突變 → 考慮CNI治療
↓
步驟5:如果CNI也失敗
↓
可能是未識別的基因形式或真正的抗治療免疫疾病
↓
進一步免疫抑制是徒勞且危險的
↓
專注:血壓控制、RAS阻斷、水腫管理、抗凝(如需要)
Meyrier的核心信息是:首先,不傷害。
「我們對FSGS治療的熱情常常導致過度治療,」他警告。「類固醇可以導致骨折、感染、糖尿病。CNI可以導致腎毒性。細胞毒性藥物可以導致不孕和癌症。如果患者的FSGS是基因性的——這種情況比我們以前認識的更常見——所有這些毒性都是徒勞的。」
他強調對類固醇和CNI雙重抗性的患者,最明智的策略通常是:
- 停止免疫抑制
- 最大化ACE抑制劑或ARB劑量(這些藥物可以降低腎小球內壓力,減少蛋白尿,延緩進展)
- 控制血壓
- 管理水腫和其他腎病症候群併發症
- 接受保守治療的概念
Meyrier的智慧為基因檢測革命鋪平了道路。當NPHS2、INF2和其他基因被發現時,他的框架提供了整合這些信息的臨床路徑。
第十三章:新曙光(2023)
數十年來,FSGS治療的核心問題是:除了免疫抑制,我們還有什麼武器?
答案終於在2023年出現。
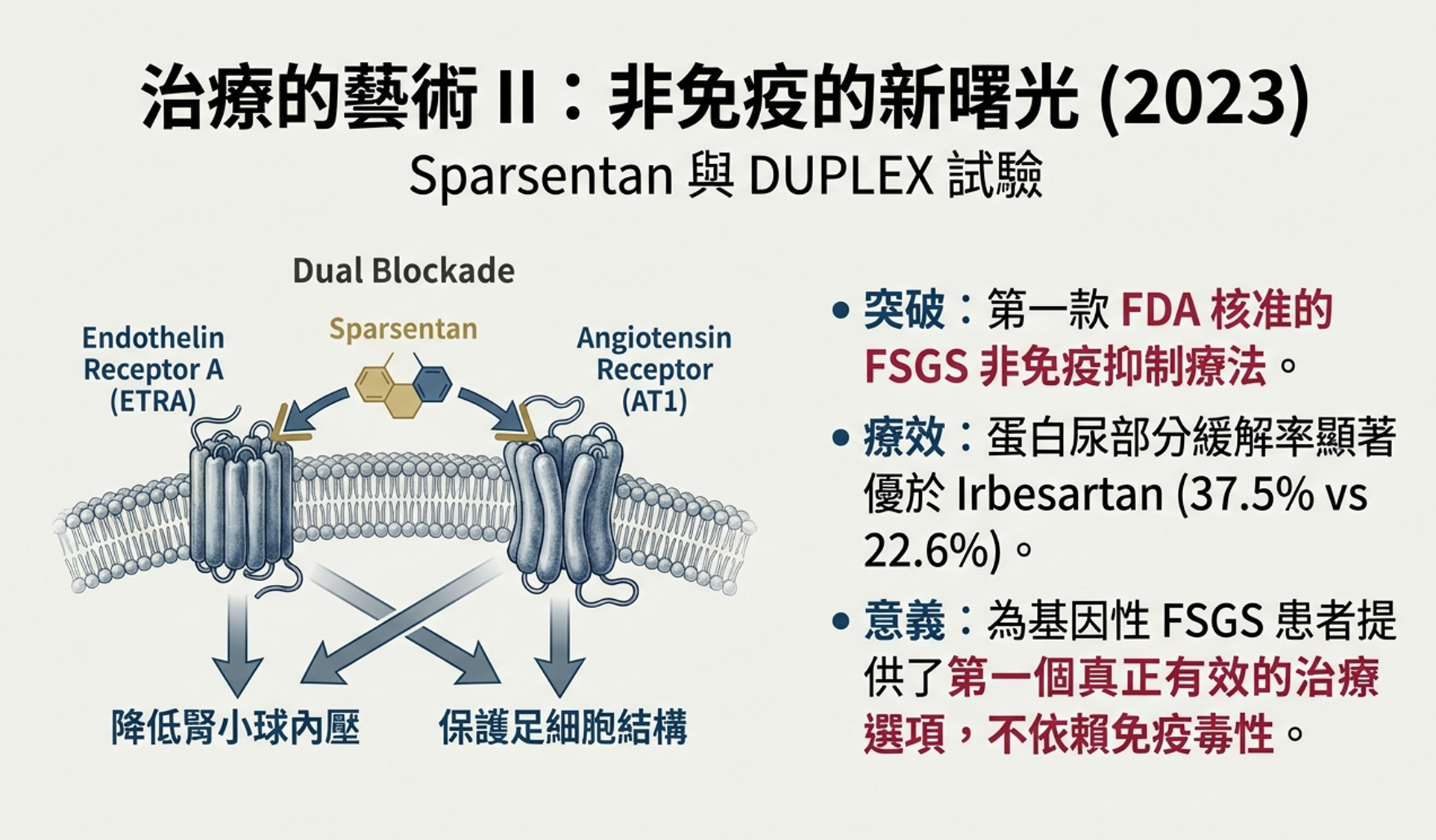
Sparsentan是一種新型藥物,它同時阻斷兩個受體:內皮素受體A(ETRA)和血管緊張素II受體。它不是免疫抑制劑——它的作用機制是血流動力學和足細胞保護。
內皮素是一種強效血管收縮劑,在蛋白尿腎臟病中表達增加。它不僅收縮血管,還直接作用於足細胞,損害其功能。阻斷內皮素受體可以保護足細胞並降低腎小球內壓力。
DUPLEX試驗是一項三期、隨機、雙盲試驗,比較Sparsentan和Irbesartan(一種單純的ARB)在FSGS患者中的療效。Irbesartan是公認的腎臟保護藥物,所以這是一個高門檻的比較——Sparsentan需要證明它比已知有效的治療更好。
2023年12月,結果發表在《新英格蘭醫學雜誌》:
蛋白尿終點:
- Sparsentan組:37.5%的患者達到部分緩解
- Irbesartan組:22.6%達到部分緩解
Sparsentan顯著減少蛋白尿——這是FSGS進展的關鍵驅動因素。
eGFR斜率(腎功能下降速度)的結果更為複雜。兩組之間的差異未達統計學意義(p=0.7491)。這意味著雖然Sparsentan減少了蛋白尿,但在這項試驗的隨訪期內,它尚未轉化為可測量的腎功能保護優勢。
儘管如此,美國FDA基於蛋白尿終點授予Sparsentan加速批准,使其成為第一個專門獲批用於FSGS的非免疫抑制藥物。
DUPLEX的意義超越了單一藥物:
- 證明非免疫途徑可行:Sparsentan證明可以不依賴免疫抑制來治療FSGS。這對基因性FSGS患者尤其重要——他們終於有了一種合理的治療選擇。
- 驗證內皮素生物學:多年來關於內皮素在腎臟病中作用的基礎研究得到臨床驗證。
- 聯合治療可能性:理論上,Sparsentan可以與免疫抑制聯合使用,提供互補的保護機制。
- 跨疾病適用性:Sparsentan在IgA腎病的PROTECT試驗中也顯示療效,表明它可能是一種通用的腎臟保護藥物。
Michelle Rheault,DUPLEX試驗的首席研究者,在發表後說:「這不是FSGS治療的終點,但它是一個重要的轉折點。我們終於有了免疫抑制之外的選項。」
第五部:移植的賭博
第十四章:復發的恐懼
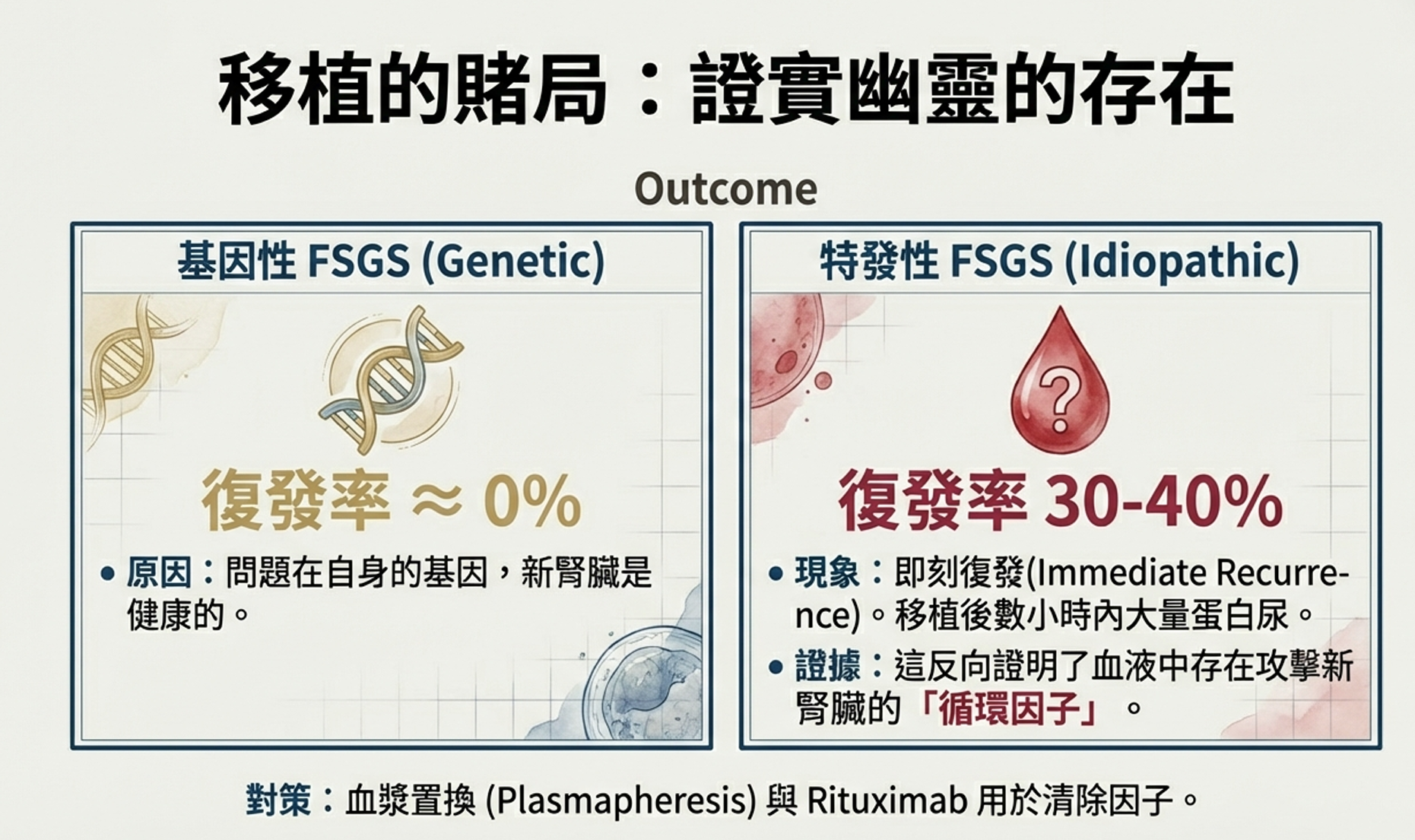
對於進展到末期腎病的FSGS患者,腎臟移植提供了擺脫透析的希望。
但移植帶來一個獨特的威脅:FSGS可能在新腎臟中復發。
想像一下:一名患者在透析上煎熬了五年,終於等到了一個匹配的腎臟。手術成功,新腎臟開始產尿。但就在幾小時後,尿液中出現大量蛋白質。活檢顯示:足突融合,腎小球損傷正在重演。
這是最殘酷的轉折——原本是重生的時刻,變成了噩夢的重演。
復發率有多高?
2013年,荷蘭的Rutger Maas團隊發表了一項回顧性研究,分析94名接受移植的FSGS患者。他們識別出復發的風險因素:
- 診斷時嚴重低白蛋白血症
- 移植前大量蛋白尿
- 原生腎快速進展至腎衰竭
但最引人注目的發現是:基因性FSGS患者幾乎從不復發。
在他們的隊列中,有確認基因突變的患者復發率是0%,而「特發性」患者的復發率是42%。
這個發現具有深刻的臨床意義:
- 基因檢測可以指導移植決策:對於攜帶NPHS2、INF2等突變的患者,移植是極好的選擇,因為復發風險極低
- 特發性患者需要密切監測:他們面臨顯著的復發風險,可能需要預防性血漿置換或其他策略
- 復發機制得到確認:如果基因性患者不復發,這意味著復發是由循環因子驅動的——這是Shalhoub假說的最強臨床證據
2019年,國際多中心TANGO研究提供了更大規模的數據。這項研究涉及17個移植中心,篩選了超過11,000名受者以識別FSGS病例。
關鍵發現:
- 特發性FSGS的復發率:32%
- 復發使移植腎喪失風險增加五倍
- 接受強化治療(血漿置換、Rituximab)的復發患者中,只有57%達到緩解
- 因復發導致的既往移植腎喪失,在後續移植中攜帶80-100%的復發風險
TANGO是FSGS復發的「弗雷明翰研究」——它提供了決定性的流行病學基準,影響全球的移植實踐。
第十五章:Rituximab的希望
面對移植後復發,腎臟科醫師們嘗試了各種挽救治療。
血漿置換是第一線選擇——通過移除血漿中的蛋白質(包括假設的循環因子),可以暫時減少蛋白尿。但這是一種繁瑣的治療,需要頻繁的透析樣療程,而且效果通常不持久。
2014年,義大利Mario Negri研究所的Piero Ruggenenti報告了一個新的選擇:Rituximab——一種抗CD20單克隆抗體,可以耗竭B細胞。
Rituximab最初開發用於治療淋巴瘤,後來被發現對自身免疫性疾病有效。它在類風濕性關節炎、系統性紅斑狼瘡和一些腎小球腎炎中已經證明了價值。
Ruggenenti的團隊報告,在血漿置換失敗的復發患者中,Rituximab可以誘導緩解。雖然這不是隨機試驗,但嚴格的觀察數據令人印象深刻。
為什麼B細胞耗竭會對FSGS有效?
有幾種假說:
- 循環因子可能是B細胞產物:也許那個難以捉摸的通透因子是一種抗體或B細胞分泌的分子
- Rituximab可能直接作用於足細胞:一些研究表明Rituximab可以結合足細胞上的SMPDL-3b蛋白,穩定細胞骨架。這將是一種「off-target」但有益的效應
- 廣泛的免疫調節:B細胞耗竭可能影響整個免疫網絡,包括T細胞功能
無論機制如何,Rituximab現在被納入許多移植中心的復發治療方案中。它不是萬靈藥——許多患者仍然對它無反應——但它為一些絕望的病例提供了希望。
尾聲:未完的旅程
2024年,當我們回顧FSGS研究的半世紀歷程,看到的是一幅壯觀的科學史詩。
從Shalhoub在1974年提出的模糊假說,到今天精確的分子分類;從「類脂質腎病」這個含糊的名稱,到數十種已識別的基因疾病;從只有類固醇這一種武器,到CNIs、Rituximab、Sparsentan組成的治療武器庫——進步是巨大的。
但未完成的任務同樣艱鉅。
幽靈因子仍在逃亡。儘管suPAR和B7-1的假說已經褪色,原發性FSGS中循環因子的概念仍然有力——移植後即刻復發的臨床現象無法用其他方式解釋。某種未知的分子正在血液中遊蕩,攻擊腎小球。識別它將是下一個重大突破。
精準醫學仍是目標。理想的未來是:每一位FSGS患者都接受全面的基因檢測和生物標記物評估;基於其特定的分子缺陷,制定個人化的治療方案;基因性患者接受足細胞保護和腎臟保護治療(或基因治療);免疫性患者接受靶向免疫治療;沒有人再接受毫無希望的免疫抑制毒性。
我們還沒有到達那裡,但路徑已經清晰。
APOL1抑制劑正在路上。多家製藥公司正在開發針對APOL1的藥物,進入臨床試驗。如果成功,這將是第一個針對非裔人群特定遺傳風險的精準治療,有望減少這一人群中腎臟病的巨大負擔。
基因治療曙光乍現。對於單基因形式的FSGS,基因治療在理論上是可行的——替換缺陷的NPHS2或INF2基因,讓足細胞恢復正常功能。這在技術上極具挑戰性(如何將基因遞送到足細胞?如何確保長期表達?),但不再是科幻。
回到本文開頭的那個十歲男孩——眼睛腫脹、腹部膨脹、尿液充滿蛋白質。
如果他在1974年發病,醫師能做的只是給他類固醇,然後希望最好的結果。如果類固醇無效,他可能在青春期之前就需要透析。
如果他在2024年發病,他的旅程會非常不同:
- 首先,全面的家族史和基因檢測,識別任何可能的遺傳原因
- 如果發現NPHS2或其他突變,避免無效的免疫抑制,直接轉向保守治療和足細胞保護藥物
- 如果是特發性,嘗試類固醇,然後是CNI,密切監測反應
- 無論病因如何,都使用Sparsentan或其他腎臟保護藥物
- 如果進展到需要移植,基於復發風險進行個人化的圍手術期管理
- 如果攜帶APOL1高風險基因型,可能有機會參加新型治療的臨床試驗
這就是五十年科學進步的意義:不是治癒所有人,但是給每一位患者更好的機會,更精準的治療,更少的不必要傷害。
Shalhoub在1974年描繪的那個幽靈——循環通透因子——仍然若隱若現。也許它將在下一篇里程碑論文中被捕獲。也許它將被證明是一個美麗但錯誤的假說。無論如何,對它的追尋已經推動了整個領域前進,揭示了足細胞的秘密,改變了無數患者的命運。
這就是醫學研究的本質:不是單一的勝利時刻,而是一代又一代科學家的接力賽,每一棒都將火炬傳得更遠一些。
FSGS的故事還沒有結束。最精彩的篇章,可能正在被書寫。
謹以此文獻給所有與FSGS搏鬥的患者、家屬,以及為理解和治療這種疾病而不懈努力的臨床醫師和科學家。
附錄:時間線
| 年代 | 里程碑 |
|---|---|
| 1974 | Shalhoub提出T細胞假說和循環因子概念 |
| 1984 | Rao描述HIV相關腎病(HIVAN) |
| 1998 | Kestilä發現nephrin基因(NPHS1) |
| 1999 | Cattran的環孢素隨機試驗 |
| 2000 | Boute發現podocin基因(NPHS2) |
| 2000 | Kaplan發現α-actinin-4基因(ACTN4) |
| 2004 | D'Agati發表哥倫比亞分類 |
| 2005 | Winn發現TRPC6基因 |
| 2010 | Brown發現INF2基因 |
| 2010 | Genovese發現APOL1風險變異 |
| 2011 | Wei提出suPAR假說 |
| 2011 | Gipson發表FSGS-CT試驗結果 |
| 2013 | Yu提出B7-1/Abatacept假說 |
| 2015 | Spinale反駁suPAR作為FSGS特異性因子 |
| 2016 | Greenbaum的Abatacept試驗失敗 |
| 2019 | TANGO研究發表移植後復發數據 |
| 2023 | DUPLEX試驗證明Sparsentan療效 |
核心概念詞彙表
| 術語 | 定義 |
|---|---|
| 足細胞 | 覆蓋腎小球毛細血管的特化上皮細胞,是過濾屏障的關鍵組件 |
| 裂孔隔膜 | 足細胞足突之間的精巧結構,含nephrin、podocin等蛋白 |
| 循環通透因子 | 假設存在的血漿分子,可增加腎小球通透性 |
| 哥倫比亞分類 | 2004年提出的FSGS五種病理變異型分類系統 |
| APOL1 | 載脂蛋白L1基因,其風險變異與非裔人群腎臟病顯著相關 |
| 類固醇抗性 | 對類固醇治療無反應,常提示基因性或嚴重免疫性疾病 |
| CNI | 鈣調神經磷酸酶抑制劑(環孢素、他克莫司) |
| 移植後復發 | FSGS在移植腎中重新出現,約1/3特發性患者發生 |
參考文獻
本文基於以下20篇里程碑論文及其相關文獻撰寫:
- Shalhoub RJ. Lancet. 1974
- Rao TK et al. N Engl J Med. 1984
- D'Agati VD et al. Am J Kidney Dis. 2004
- Kestilä M et al. Mol Cell. 1998
- Boute N et al. Nat Genet. 2000
- Kaplan JM et al. Nat Genet. 2000
- Winn MP et al. Science. 2005
- Brown EJ et al. Nat Genet. 2010
- Genovese G et al. Science. 2010
- Kopp JB et al. J Am Soc Nephrol. 2011
- Wei C et al. Nat Med. 2011
- Spinale JM et al. Kidney Int. 2015
- Yu CC et al. N Engl J Med. 2013
- Cattran DC et al. Kidney Int. 1999
- Gipson DS et al. Kidney Int. 2011
- Meyrier A. Expert Opin Pharmacother. 2009
- Rheault MN et al. N Engl J Med. 2023
- Maas RJ et al. BMC Nephrol. 2013
- Uffing A et al. Clin J Am Soc Nephrol. 2019
- Ruggenenti P et al. J Am Soc Nephrol. 2014